Jill Mesirov & Eric
Lander talk with ScienceWatch.com and answer a few
questions about this month's Fast Moving Front in the field
of Computer Science. The authors have also sent along
images of their work.
Article: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide
expression profiles
Authors: Subramanian, A;Tamayo, P;Mootha, VK;Mukherjee,
S;Ebert, BL;Gillette, MA;Paulovich, A;Pomeroy, SL;Golub,
TR;Lander,
ES;Mesirov,
JP
Journal: PROC NAT ACAD SCI USA, 102 (43): 15545-15550 OCT
25 2005
Addresses: MIT, Broad Inst, 320 Charles St, Cambridge, MA
02141 USA.
MIT, Broad Inst, Cambridge, MA 02141 USA.
Harvard Univ, Cambridge, MA 02141 USA.
Harvard Univ, Sch Med, Dept Syst Biol, Boston, MA 02446
USA.
Duke Univ, Inst Genome Sci & Policy, Ctr
Interdisciplinary Engn Med & Appl Sci, Durham, NC 27708
USA.
Why do you think your paper is highly
cited?
The paper described the methodology, and announced the availability of a
software implementation, for a new paradigm for extracting biological
insights from genome-wide transcription profiles which was first introduced
in Mootha, VK et al. "PGC-1alpha responsive genes involved in
oxidative phosphorylation are coordinately downregulated in human
diabetes," Nat. Genet. 34:267-73, 2003. Until that time, the
common approach to analyzing such data was at the level of individual
genes.
Gene Set Enrichment Analysis (GSEA) derives its power by considering sets
of genes that correspond to a biological process, a chromosomal location,
or have a common regulatory pattern. GSEA addresses a number of problems
associated with the single-gene approach to interpreting expression data.
It enables the identification of subtle but relevant biological differences
in phenotypes, aids in the interpretation of data through the use of
annotated sets in the accompanying Molecular Signatures Database (MSigDB)
and, importantly, its results tend to be more reproducible across data sets
and technology platforms.
There are thousands of users of the GSEA software and the related,
annotated MSigDB. It has become the method of choice for the initial
analysis of global expression data much as BLAST (Basic Local Alignment
Search Tool) is commonly used for sequence data.
Would you summarize the significance of your paper in
layman's terms?
The method and the software implementation help biomedical researchers to
identify the underlying biological processes associated with cellular
states. Through this approach they may gain insights into the mechanisms of
disease.
How did you become involved in this research and were
there any particular problems encountered along the way?
This research began with the work described in the Mootha, VK et al.
Nat. Genet. paper that sought to identify significant differences
between samples of patients with type II diabetes and those with normal
glucose tolerance. While single-gene approaches showed no significant
difference between the two, GSEA was able to identify a set of genes
associated with the oxidative phosphorylation pathway that was
significantly down-regulated in the diabetic patients.
The problems we faced were associated with generalizing the method for use
with other data sets. In particular, the initial approach identified sets
of genes that exhibited any nonrandom behavior, while the method described
in the cited paper detects sets of genes that are coordinately up- or
down-regulated.
Given expression data for samples of two different cellular states or
phenotypes, GSEA starts by ranking the genes according to their correlation
with those states. The goal of GSEA is to determine whether a set of genes
corresponding to a biological pathway or process or a cytogenetic band are
randomly distributed throughout the list or over-represented at the top or
bottom. Sets related to the distinction between the states should tend to
show the latter behavior.
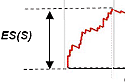
An "enrichment score" for the gene set is calculated by walking down the
list of genes and increasing the score, by an amount proportional to the
gene's correlation with the phenotype of interest, if gene is in the set,
and decreasing the score if gene is not in the set. Significance of the
score is estimated by a permutation test. The use of the weighted step
identifies sets of genes that are over-represented at the top (or bottom)
of a gene list, rather than other non-random behaviors as would be the case
with the Kolmogorov-Smirnov statistic.
Eric S. Lander, Ph.D.
Director
Broad Institute of Massachusetts Institute of Technology and Harvard
Cambridge, MA, USA
Jill P. Mesirov, Ph.D.
Associate Director, Chief Informatics Officer
Director, Computational Biology and Bioinformatics
Broad Institute of Massachusetts Institute of Technology and Harvard
Cambridge, MA, USA