J. Evan Sadler talks with
ScienceWatch.com and answers a few questions about
this month's Fast Moving Front in the field of Clinical
Medicine. The author has also sent along images of
their work.
Article: Update on the pathophysiology and
classification of von Willebrand disease: a report of the
Subcommittee on von Willebrand Factor
Authors: Sadler,
JE, et. al
J THROMB HAEMOST, 4 (10): 2103-2114 OCT 2006
Washington Univ, Sch Med, Howard Hughes Med Inst, 660 S
Euclid Ave,Box 8022, St Louis, MO 63110 USA.\nWashington
Univ, Sch Med, Howard Hughes Med Inst, St Louis, MO 63110
USA.
Lab Assoc Prof Arndt & Partners, Hamburg,
Germany.
Leiden Univ, Med Ctr, Dept Haematol, Leiden,
Netherlands.
Westmead Hosp, ICPMR, Westmead, NSW 2145, Australia.
Birmingham Childrens Hosp NHS Trust, Dept Haematol,
Birmingham, W Midlands, England.
(addresses have been truncated)
Why do you think your paper is highly
cited?
I think our paper is highly cited because it provides a relatively simple
and useful framework to organize a complex mass of biochemical and clinical
data. von Willebrand disease (VWD) is a bleeding disorder caused by
inherited defects in von Willebrand factor (VWF), which is an enormous
multimeric blood protein that is necessary for hemostasis.
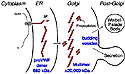
VWF is assembled (Figure 1) within endothelial cells from identical
subunits that first form dimers in the endoplasmic reticulum "tail-to-tail"
through disulfide bonds between C-terminal domains. These dimers are
transported to the Golgi, where they form multimers that are linked
"head-to-head" by disulfide bonds between N-terminal D3 domains.
The finished multimers are stored in densely packed tubular arrays within
morphologically unusual vesicles called Weibel-Palade bodies, from which
they are secreted into the blood. VWF multimers perform their hemostatic
function by binding to exposed connective tissue and to certain platelet
membrane glycoproteins, thereby facilitating the adhesion of platelets at
sites of vascular injury. VWF also binds blood clotting factor VIII and
stabilizes it in the circulation.
Biosynthesis
of VWF. The VWF
precursor is
translocated...
Classification
of
VWD...
Mutations
in VWD type 2. The VWF
precursor consists of a
sig...
The complicated biosynthesis and multiple binding functions of VWF can be
disrupted by mutations to yield many disease phenotypes that can be
challenging to diagnose and treat appropriately. The classification of VWD
that we proposed (Figure 2) condenses this variety into a manageable set of
categories, providing a common language to describe and analyze what we
know.
Does it describe a new discovery, methodology, or
synthesis of knowledge?
The classification recognizes a small number of different pathophysiologic
mechanisms which disrupt VWF biosynthesis, structure, or function. It's
intended to be simple, to rely on widely available laboratory tests, and to
correlate with important clinical characteristics.
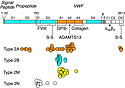
There is a reasonably good relationship between VWF genotype and VWD
phenotype, especially for the qualitative or type 2 variants of VWD (Figure
3). For example, multimer assembly requires the C-terminal CK domains for
dimerization in the endoplasmic reticulum, and both the propeptide and D3
domains for final assembly in the Golgi.
Mutations in any of these regions cause the loss of hemostatically
effective large VWF multimers, which is characteristic of VWD type 2A.
Gain-of-function mutations in the A1 domain that cause spontaneous binding
of VWF to platelets also cause VWD type 2B, which is usually associated
with thrombocytopenia. Loss-of-function mutations that impair platelet or
collagen binding cause VWD type 2M.
One of my favorite variants is VWD type 2N, which is caused by mutations in
or near the factor VIII binding site. These patients have very low factor
VIII levels and sometimes are misdiagnosed as having hemophilia A. In fact,
their factor VIII gene is normal and they simply clear their endogenous
factor VIII too quickly. Distinguishing hemophilia from VWD type 2N has
important implications for treatment. Genetic counseling also differs
because hemophilia A is an X-linked disorder, whereas VWD type 2N is
autosomal recessive.
Would you summarize the significance of your paper
in layman's terms?
VWD appears to be the most common inherited bleeding disorder, and it is
caused by many different kinds of defects in VWF. As a consequence of this
variability, patients with different types of VWD vary considerably in the
severity of bleeding symptoms and require different treatments. The paper
describes a way to distinguish several types of VWD so that, in most cases,
we can choose the best treatment to stop or prevent bleeding for our
patients.
How did you become involved in this research and
were any particular problems encountered along the way?
About 25 years ago, several research groups around the world, including
mine, cloned the VWF gene and began characterizing the mutations that cause
VWD. Within a few years I think we all realized that the wealth of new
molecular data offered an opportunity to take a fresh look at how VWD was
diagnosed and classified, which we did first in 1994 and, most recently, in
2006.
The Scientific and Standardization Committee of the International Society
on Thrombosis and Hemostasis has provided a perfect organizational
structure to accomplish this task through their Subcommittee on von
Willebrand Factor, which includes active basic and clinical investigators
with broad international representation.
I would say that our major problem was simply sorting through the vast
literature on VWD and distilling it into a relatively concise framework
that did not unintentionally misrepresent the state of the science. I have
been very impressed by the world community of researchers that study
bleeding disorders, which has been unfailingly collegial, dedicated, and
willing to work. Achieving consensus was relatively easy, although
coordinating authors from so many countries was a challenge and our
manuscript required approximately two years to write.
Where do you see your research leading in the
future?
Clearly there are gaps in our knowledge, and no classification we can
devise today will be perfect. For example, patients with any particular
mutation don't necessarily have the same severity of bleeding symptoms.
There must be other factors in a person's genetic makeup, environment, or
life history that influences how they respond to mutations in the VWF gene.
One of our biggest challenges is to understand these factors and the
interplay between them, so that we can understand the relationship between
VWF genotype and disease phenotype. Many of my coauthors are actively
engaged in translational or clinical research to address this problem.
My laboratory is focusing on biochemical studies of how VWF multimers are
assembled and how they function. Together, these approaches should lead to
better diagnostic tests and treatments that are tailored for the unique
needs of each patient.
Do you foresee any social or political implications
for your research?
I can give an example that relates to women's health, which is a social and
political issue worldwide. Excessive menstrual bleeding is especially
common in VWD, and is a significant cause of disability and iron-deficiency
anemia. In fact, most patients with symptomatic VWD are women. Research
summarized in our classification paper has led to an increased awareness of
VWD as a significant medical problem, particularly for women, which I hope
will result in the wider availability of treatments to improve their
quality of life.
J. Evan Sadler, M.D., Ph.D.
Professor of Medicine
Chief, Hematology Division
Professor of Biochemistry & Molecular Biophysics
Washington University School of Medicine
St. Louis, MO, USA Web
KEYWORDS: ADAMTS-13; CLASSIFICATION; PATHOPHYSIOLOGY;
VON WILLEBRAND DISEASE.